Figure 3 and 4 - DE / GSEA PanCan
Waldron Lab
May 11, 2020
Source:vignettes/Figures3n4_GSEA_PanCan.Rmd
Figures3n4_GSEA_PanCan.RmdObtain ACC data from curatedTCGAData:
data("diseaseCodes") head(avail <- diseaseCodes[ diseaseCodes[["Available"]] == "Yes", "Study.Abbreviation"])
## [1] "ACC" "BLCA" "BRCA" "CESC" "CHOL" "COAD"Download and reshape data
Some datasets were missing normals and we suspected that there were different datasets being produced by RTCGAToolbox. Upon further inspection, TCGA provides ‘illuminaga’ and ‘illumina HiSeq’ datasets.
Note. First, download RTCGAToolbox from Bioc-devel and obtain correlations for samples that are in both platforms in “COAD”, “READ”, and “UCEC”.
dl <- c("COAD", "READ", "UCEC") redl <- lapply(setNames(dl, dl), function(x) { tx <- getFirehoseData(x, RNASeq2GeneNorm = TRUE, clinical = FALSE) txl <- biocExtract(tx, "RNASeq2GeneNorm") sameSamps <- Reduce(intersect, lapply(txl, colnames)) # correlation between platforms 'illuminaga' and 'illuminahiseq' message(x) datm <- do.call(cbind.data.frame, lapply(txl, function(g) { assay(g[,sameSamps]) }) ) if (length(datm)) print(cor(datm[,1], datm[,2])) newassay <- cbind(assay(txl[[1]]), assay(txl[[2]])) newassay <- data.matrix(newassay[, !duplicated(colnames(newassay))]) newmeta <- c(metadata(txl[[1]]), metadata(txl[[2]])) newColData <- DataFrame(row.names = colnames(newassay)) SummarizedExperiment(assays = list(TPM = newassay), metadata = newmeta, colData = newColData) } )
## [1] 0.927677
## [1] 0.8698399exper <- as(redl, "ExperimentList") sampleTables(MultiAssayExperiment(exper))
## $COAD
##
## 01 02 06 11
## 457 1 1 41
##
## $READ
##
## 01 02 11
## 166 1 10
##
## $UCEC
##
## 01 02 11
## 545 1 35Create a pseudo clinical DataFrame for new samples
allSamps <- unlist(colnames(exper)) clinphone <- DataFrame(patientID = TCGAbarcode(allSamps)) rownames(clinphone) <- clinphone[["patientID"]] sum(duplicated(rownames(clinphone)))
## [1] 77## [1] 77 clinphone <- clinphone[!duplicated(rownames(clinphone)), , drop = FALSE]
Combine cancer datasets into one MultiAssayExperiment:
outAssays <- c("COAD_RNASeq2GeneNorm-20160128", "READ_RNASeq2GeneNorm-20160128", "UCEC_RNASeq2GeneNorm-20160128") core <- exper[ c("COAD", "READ", "UCEC") ] names(core) <- outAssays mapper <- generateMap(core, clinphone, idConverter = TCGAbarcode) coremae <- MultiAssayExperiment( experiments = core, sampleMap = mapper, colData = clinphone )
PanCan RNASeq2*
Obtain the RNAseq data from curatedTCGAData:
rnacomp <- curatedTCGAData("*", "RNASeq2*", FALSE) rnacomp
## A MultiAssayExperiment object of 33 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 33:
## [1] ACC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 79 columns
## [2] BLCA_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 427 columns
## [3] BRCA_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 1212 columns
## [4] CESC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 309 columns
## [5] CHOL_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 45 columns
## [6] COAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 191 columns
## [7] DLBC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 48 columns
## [8] ESCA_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 196 columns
## [9] GBM_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 166 columns
## [10] HNSC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 566 columns
## [11] KICH_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 91 columns
## [12] KIRC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 606 columns
## [13] KIRP_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 323 columns
## [14] LAML_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 173 columns
## [15] LGG_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 530 columns
## [16] LIHC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 423 columns
## [17] LUAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 576 columns
## [18] LUSC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 552 columns
## [19] MESO_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 87 columns
## [20] OV_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 307 columns
## [21] PAAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 183 columns
## [22] PCPG_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 187 columns
## [23] PRAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 550 columns
## [24] READ_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 72 columns
## [25] SARC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 265 columns
## [26] SKCM_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 473 columns
## [27] STAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 450 columns
## [28] TGCT_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 139 columns
## [29] THCA_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 568 columns
## [30] THYM_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 122 columns
## [31] UCEC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 380 columns
## [32] UCS_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 57 columns
## [33] UVM_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 80 columns
## Features:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DFrame
## sampleMap() - the sample availability DFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DFrame
## assays() - convert ExperimentList to a SimpleList of matricesReplace above processed datasets in the full RNASeq MultiAssayExperiment:
rnacopy <- rnacomp rnacomp <- rnacomp[, , !names(rnacomp) %in% outAssays]
## harmonizing input:
## removing 643 sampleMap rows not in names(experiments)
## removing 633 colData rownames not in sampleMap 'primary'rnacomp <- c(rnacomp, coremae)
Here we check for the samples of interest using sampleTables:
sampleTables(rnacomp)
## $`ACC_RNASeq2GeneNorm-20160128`
##
## 01
## 79
##
## $`BLCA_RNASeq2GeneNorm-20160128`
##
## 01 11
## 408 19
##
## $`BRCA_RNASeq2GeneNorm-20160128`
##
## 01 06 11
## 1093 7 112
##
## $`CESC_RNASeq2GeneNorm-20160128`
##
## 01 06 11
## 304 2 3
##
## $`CHOL_RNASeq2GeneNorm-20160128`
##
## 01 11
## 36 9
##
## $`DLBC_RNASeq2GeneNorm-20160128`
##
## 01
## 48
##
## $`ESCA_RNASeq2GeneNorm-20160128`
##
## 01 06 11
## 184 1 11
##
## $`GBM_RNASeq2GeneNorm-20160128`
##
## 01 02
## 153 13
##
## $`HNSC_RNASeq2GeneNorm-20160128`
##
## 01 06 11
## 520 2 44
##
## $`KICH_RNASeq2GeneNorm-20160128`
##
## 01 11
## 66 25
##
## $`KIRC_RNASeq2GeneNorm-20160128`
##
## 01 05 11
## 533 1 72
##
## $`KIRP_RNASeq2GeneNorm-20160128`
##
## 01 05 11
## 290 1 32
##
## $`LAML_RNASeq2GeneNorm-20160128`
##
## 03
## 173
##
## $`LGG_RNASeq2GeneNorm-20160128`
##
## 01 02
## 516 14
##
## $`LIHC_RNASeq2GeneNorm-20160128`
##
## 01 02 11
## 371 2 50
##
## $`LUAD_RNASeq2GeneNorm-20160128`
##
## 01 02 11
## 515 2 59
##
## $`LUSC_RNASeq2GeneNorm-20160128`
##
## 01 11
## 501 51
##
## $`MESO_RNASeq2GeneNorm-20160128`
##
## 01
## 87
##
## $`OV_RNASeq2GeneNorm-20160128`
##
## 01 02
## 303 4
##
## $`PAAD_RNASeq2GeneNorm-20160128`
##
## 01 06 11
## 178 1 4
##
## $`PCPG_RNASeq2GeneNorm-20160128`
##
## 01 05 06 11
## 179 3 2 3
##
## $`PRAD_RNASeq2GeneNorm-20160128`
##
## 01 06 11
## 497 1 52
##
## $`SARC_RNASeq2GeneNorm-20160128`
##
## 01 02 06 11
## 259 3 1 2
##
## $`SKCM_RNASeq2GeneNorm-20160128`
##
## 01 06 07 11
## 103 368 1 1
##
## $`STAD_RNASeq2GeneNorm-20160128`
##
## 01 11
## 415 35
##
## $`TGCT_RNASeq2GeneNorm-20160128`
##
## 01 05
## 134 5
##
## $`THCA_RNASeq2GeneNorm-20160128`
##
## 01 06 11
## 501 8 59
##
## $`THYM_RNASeq2GeneNorm-20160128`
##
## 01 11
## 120 2
##
## $`UCS_RNASeq2GeneNorm-20160128`
##
## 01
## 57
##
## $`UVM_RNASeq2GeneNorm-20160128`
##
## 01
## 80
##
## $`COAD_RNASeq2GeneNorm-20160128`
##
## 01 02 06 11
## 457 1 1 41
##
## $`READ_RNASeq2GeneNorm-20160128`
##
## 01 02 11
## 166 1 10
##
## $`UCEC_RNASeq2GeneNorm-20160128`
##
## 01 02 11
## 545 1 35Which cancer codes have both tumors and normal samples?
okCA <- vapply(sampleTables(rnacomp), function(x) { all(c("01", "11") %in% names(x)) }, logical(1L) ) ( okCodes <- vapply( strsplit(names(which(okCA)), "_"), `[`, character(1L), 1L) )
## [1] "BLCA" "BRCA" "CESC" "CHOL" "ESCA" "HNSC" "KICH" "KIRC" "KIRP" "LIHC"
## [11] "LUAD" "LUSC" "PAAD" "PCPG" "PRAD" "SARC" "SKCM" "STAD" "THCA" "THYM"
## [21] "COAD" "READ" "UCEC"Assemble appropriate MultiAssayExperiment object with cancer codes that have both tumors and normals.
rnacomp2 <- rnacomp[ , , okCA]
## harmonizing input:
## removing 1666 sampleMap rows not in names(experiments)
## removing 1638 colData rownames not in sampleMap 'primary'Isolate tumor and normal samples for each cancer that have more than 10 samples of each:
selects <- TCGAsampleSelect(colnames(rnacomp2), c("01", "11")) rnacomp3 <- rnacomp2[, selects]
## harmonizing input:
## removing 410 sampleMap rows with 'colname' not in colnames of experiments
## removing 365 colData rownames not in sampleMap 'primary'enoughNorm <- vapply(sampleTables(rnacomp3), function(samp) { samp["11"] >= 10L }, logical(1L)) rnacomp4 <- rnacomp3[, , enoughNorm]
## harmonizing input:
## removing 1203 sampleMap rows not in names(experiments)
## removing 1180 colData rownames not in sampleMap 'primary'sampleTables(rnacomp4)
## $`BLCA_RNASeq2GeneNorm-20160128`
##
## 01 11
## 408 19
##
## $`BRCA_RNASeq2GeneNorm-20160128`
##
## 01 11
## 1093 112
##
## $`ESCA_RNASeq2GeneNorm-20160128`
##
## 01 11
## 184 11
##
## $`HNSC_RNASeq2GeneNorm-20160128`
##
## 01 11
## 520 44
##
## $`KICH_RNASeq2GeneNorm-20160128`
##
## 01 11
## 66 25
##
## $`KIRC_RNASeq2GeneNorm-20160128`
##
## 01 11
## 533 72
##
## $`KIRP_RNASeq2GeneNorm-20160128`
##
## 01 11
## 290 32
##
## $`LIHC_RNASeq2GeneNorm-20160128`
##
## 01 11
## 371 50
##
## $`LUAD_RNASeq2GeneNorm-20160128`
##
## 01 11
## 515 59
##
## $`LUSC_RNASeq2GeneNorm-20160128`
##
## 01 11
## 501 51
##
## $`PRAD_RNASeq2GeneNorm-20160128`
##
## 01 11
## 497 52
##
## $`STAD_RNASeq2GeneNorm-20160128`
##
## 01 11
## 415 35
##
## $`THCA_RNASeq2GeneNorm-20160128`
##
## 01 11
## 501 59
##
## $`COAD_RNASeq2GeneNorm-20160128`
##
## 01 11
## 457 41
##
## $`READ_RNASeq2GeneNorm-20160128`
##
## 01 11
## 166 10
##
## $`UCEC_RNASeq2GeneNorm-20160128`
##
## 01 11
## 545 35Create an index to annotate each of the SummarizedExperiment objects contained in the ExperimentList:
tlogic <- TCGAsampleSelect(colnames(rnacomp4), "01") sampIndx <- IntegerList(lapply(tlogic, ifelse, 1L, 0L)) naks <- mendoapply(function(x, y) { colData(x)[["GROUP"]] <- y colData(x)[["BLOCK"]] <- TCGAbarcode(rownames(colData(x))) x }, x = experiments(rnacomp4), y = sampIndx) rnacomp5 <- BiocGenerics:::replaceSlots(rnacomp4, ExperimentList = naks, check = FALSE) table(rnacomp5[[1L]]$GROUP)
##
## 0 1
## 19 408Split, create matched tumors and normals, and then recombine into a single MultiAssayExperiment:
matchlist <- lapply(setNames(seq_along(rnacomp5), names(rnacomp5)), function(idx) { as(splitAssays(rnacomp5[, , idx]), "MatchedAssayExperiment") } ) newEXPS <- ExperimentList( lapply(matchlist, function(expr) { do.call(cbind, experiments(expr)) }) ) rnacomp6 <- BiocGenerics:::replaceSlots(rnacomp5, ExperimentList = newEXPS, check = FALSE )
Save point:
saveRDS(experiments(rnacomp6), file = "../inst/data/pancan_exprs.rds") r6ex <- loadRDS(file = "../inst/data/pancan_exprs.rds")
Differential Expression analysis:
exp.list <- experiments(rnacomp6) names(exp.list) <- substring(names(exp.list), 1, 4) exp.list <- lapply(exp.list, function(se) { # if assay is matrix: assay(se) <- log(assay(se) + 1, base=2) assays(se) <- list(TPM=log(as.matrix(assay(se)) + 1, base=2)) return(se) }) exp.list <- runDE(exp.list) # for(n in names(exp.list)) saveRDS(exp.list[[n]], file = paste(n, "rds", sep="."))
Figure 3 BoxPlot
Extract genes with consistent expression changes:
wfcs <- metaFC(exp.list, max.na=3) # Heatmap top.wfcs.genes <- names(wfcs)[1:50] extractFC <- function(exp.list, top.wfcs.genes) { fcs <- vapply(exp.list, function(d) rowData(d)[top.wfcs.genes, "FC"], numeric(length(top.wfcs.genes))) rownames(fcs) <- top.wfcs.genes return(fcs) } fcs <- extractFC(exp.list, top.wfcs.genes) # pdf("log2fc.pdf", paper = "special", width = 8, height = 12) Heatmap(fcs, name="log2FC", show_row_names=TRUE)
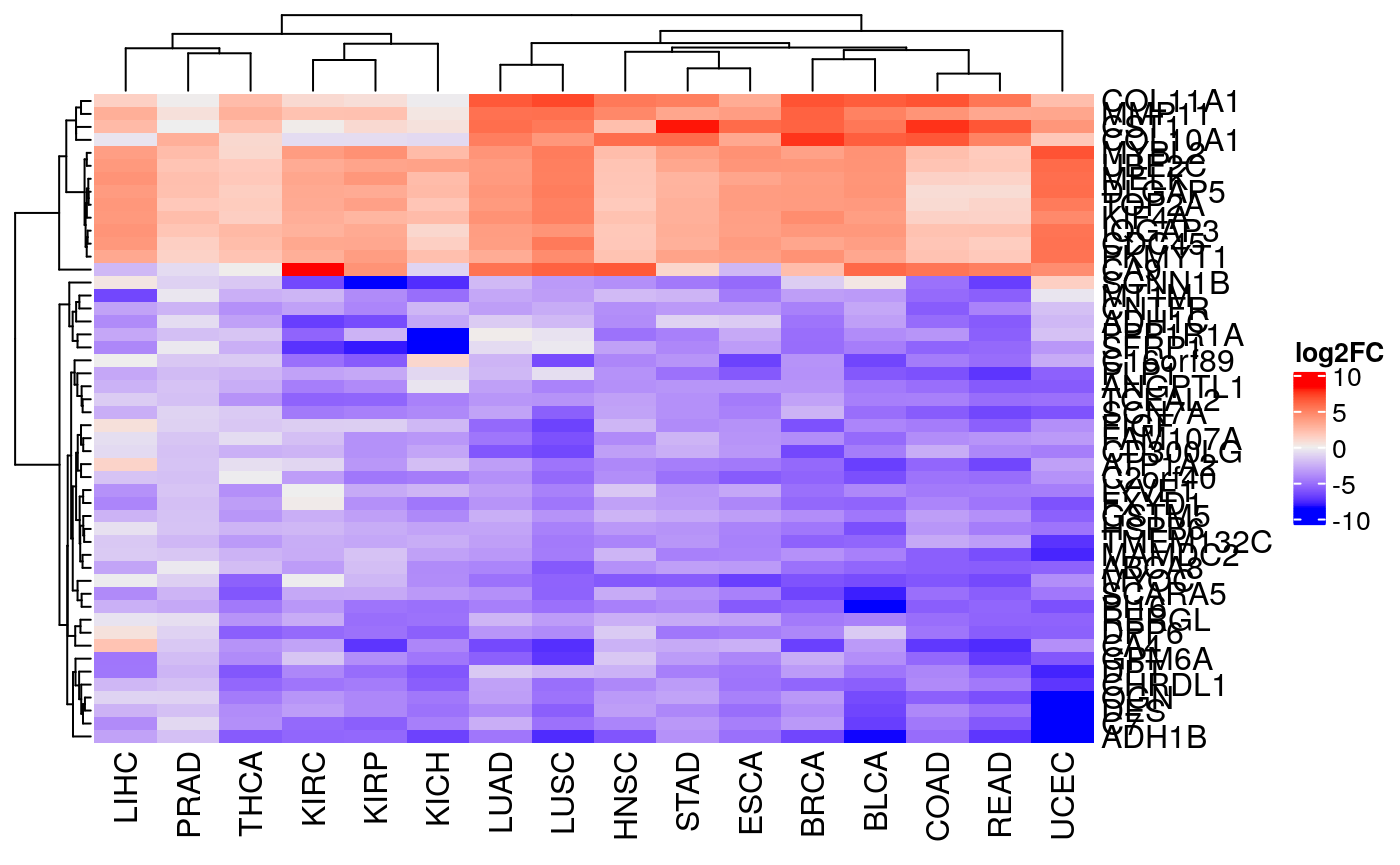
# dev.off()
Figure 3
Pan-cancer differential expression analysis
# Boxplot top.wfcs.genes <- names(c(wfcs[wfcs > 0][1:8], wfcs[wfcs < 0][1:8])) fcs <- extractFC(exp.list, top.wfcs.genes) df <- reshape2::melt(fcs) medians <- apply(fcs, 1, median, na.rm=TRUE) o <- names(sort(medians)) p <- ggboxplot(df, x = "Var1", y = "value", width = 0.8, ylab="log2 fold change", xlab="", order=o, color="Var1", add ="jitter") p <- ggpar(p, x.text.angle=45, palette = "simpsons", legend="none") gg <- p + geom_hline(yintercept=0, linetype="dashed", color = "darkgrey") + geom_vline(xintercept=8.5, linetype="dashed", color = "darkgrey") gg
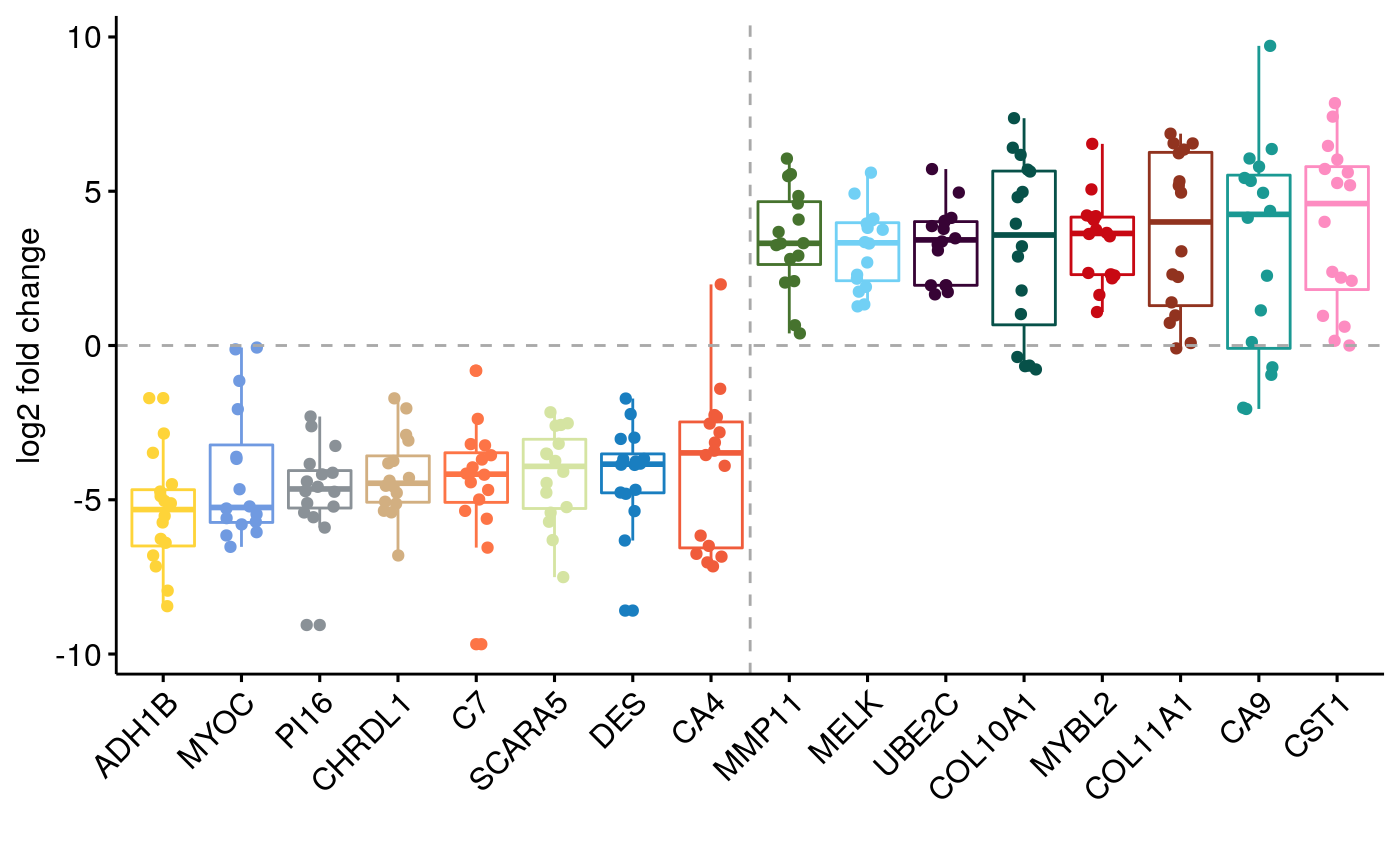
ggsaving…
ggsave("boxplot_pancan.svg", gg, device = "svg") ## pdf ggsave("boxplot_pancan.pdf", gg, device = "pdf")
GSEA
ID mapping
Obtain gene sets
go.gs <- getGenesets(org="hsa", db="go")
## ids <- vapply(names(go.gs), function(n) unlist(strsplit(n, "_"))[1], character(1)) ids <- sub(":", "", ids) extractTitle <- function(n) { spl <- unlist(strsplit(n, "_"))[-1] paste(spl, collapse = " ") } go.i2n <- vapply(names(go.gs), extractTitle, character(1)) names(go.i2n) <- ids
Execute ORA (has been pre-computed on a cluster, skip this step):
res <- runEA(exp.list, methods=c("ora", "padog"), gs=go.gs, perm=c(0, 1000), save2file=TRUE, out.dir="../inst/data")
Read pre-computed results for ORA and PADOG for GO-BP gene sets
res <- readResults(data.dir="../inst/data", data.ids=names(exp.list), methods=c("ora", "padog"), type="ranking")
Determine enrichment across datasets:
isEnriched <- function(res, pthresh=0.05, nr.top=1:15) { for(i in seq_along(res)) rownames(res[[i]]) <- res[[i]]$GENE.SET pmat <- vapply(res, function(r) r[rownames(res[[1]]), "PVAL"], res[[1]]$PVAL) rownames(pmat) <- rownames(res[[1]]) is.enriched <- pmat < pthresh rse <- rowSums(is.enriched) top.gs <- names(sort(rse, decreasing=TRUE)[nr.top]) top.mat <- is.enriched[top.gs,] mode(top.mat) <- "integer" return(top.mat) } go.ora <- isEnriched(res$ora) go.padog <- isEnriched(res$padog, nr.top=rownames(go.ora)) rownames(go.ora) <- rownames(go.padog) <- go.i2n[rownames(go.ora)] extract <- function(n, ind) paste(unlist(strsplit(n, " "))[ind], collapse = " ") rownames(go.ora)[4] <- extract(rownames(go.ora)[4], 1:2) rownames(go.ora)[10] <- extract(rownames(go.ora)[10], c(1, 3:4)) rownames(go.ora)[11] <- extract(rownames(go.ora)[11], 1:2) rownames(go.ora)[15] <- extract(rownames(go.ora)[15], 1:5) rownames(go.padog) <- rownames(go.ora)
Figure 4
Pan-cancer gene set enrichment analysis
h1 <- Heatmap(go.ora, cluster_rows=FALSE, cluster_columns=FALSE, show_row_names=FALSE, show_heatmap_legend = FALSE, col=c("white", "black"), column_title="ORA p < 0.05", row_names_gp = gpar(fontsize = 11)) h2 <- Heatmap(go.padog, cluster_rows=FALSE, cluster_columns=FALSE, show_row_names=TRUE, show_heatmap_legend = FALSE, col=c("white", "black"), column_title="PADOG p < 0.05", row_names_gp = gpar(fontsize = 11)) hl <- h1 + h2 draw(hl, ht_gap = unit(1, "cm"))
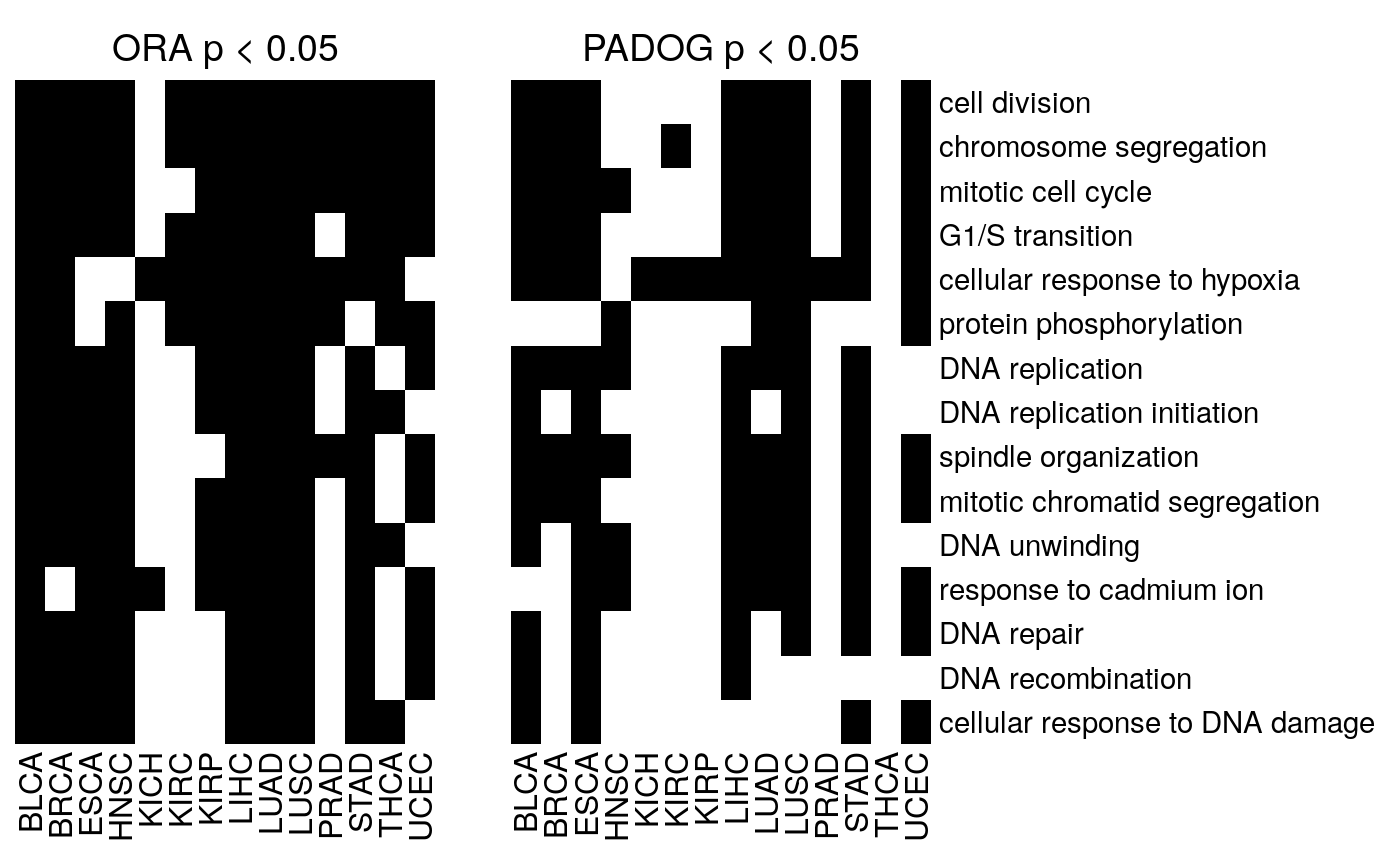
Code for saving to PDF: